All human research that takes place in NSW public health organisations (PHOs) and Specialty HealthNetworks (SHNs) must be ethically and scientifically reviewed and approved by a Human Research Ethics Committee (HREC) in accordance with the National Health Medical Research Council National Statement on Ethical Conduct in Human Research (2007), and the Policy Directive Research – Ethical & Scientific Review of Human Research in NSW Public Health Organisations.
This includes but is not limited to clinical research, clinical trials, epidemiological research, health services research, population health research, and qualitative research.
NSW supports single ethical review for studies taking place in more than one NSW PHO/SHN. NSW is also part of the National Mutual Acceptance scheme, where research taking place in public health organisations in more than one participating Australian jurisdiction needs only one* ethical and scientific review. Some exemptions apply and are available on the National Mutual Acceptance page.
Human Research Ethics Applications to a NSW or ACT Human Research Ethics Committee should be completed in the Research Ethics and Governance Information System (REGIS).
Safety notifications and reporting pathways for therapeutic goods trials
Safety notifications for therapeutic goods trials
A summary table of safety notifications to the Human Research Ethics Committee and Research Governance Officers
View safety notification summary table
- Type of event
- Who reports
- To whom
- When
- How
- Sponsor/Delegate
- The reviewing HREC (and all investigators participating in the study)
- As soon as possible and no later than 72 hours of the sponsor becoming aware of the USM
- SSI Notification Form; Sponsor’s template
- Sponsor/Delegate
- The reviewing HREC (and all investigators participating in the study)
- Within 15 days of the sponsor becoming aware of the SSI
- SSI Notification Form; Sponsor’s template
- Principal Investigator
- The RGO for the site where the event occurred
- As soon as possible and no later than 72 hours of the PI becoming aware of the SSI
- SSI Notification Form; Sponsor’s template
- Principal Investigator
- The RGO for the site where the event occurred
- Within 72 hours of the PI becoming aware of the event
- SUSAR/USADE/URSAE Notification Form
- Sponsor/Delegate
- The reviewing HREC
- As and when updates are generated
- Submitted with a cover sheet or as part of an annual progress/annual safety report
- Coordinating Principal Investigator or Sponsor/Delegate
- The reviewing HREC
- Within annual progress report sent to the HREC or aligned with the safety reporting cycles of global companies
- Annual Progress Report or sponsor’s template
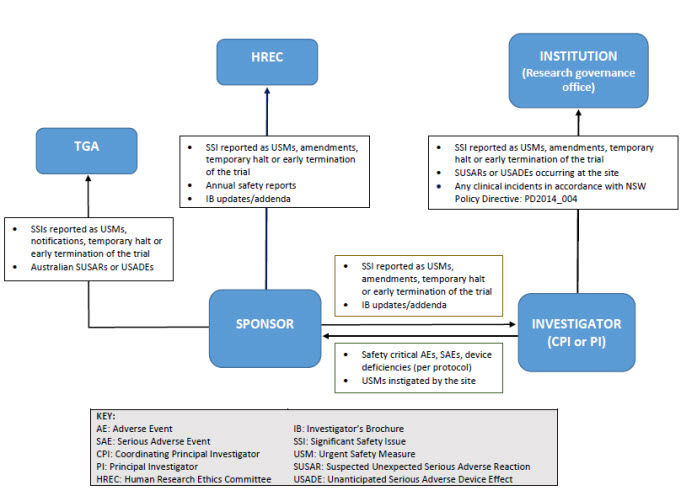
Reporting pathways for therapeutic goods trials
As illustrated below, sponsors may report directly to NSW Human Research Ethics Committee; however, they must ensure that all communications sent to the Human Research Ethics Committee adequately identify the trial and provide context in relation to the Human Research Ethics Committee’s role (e.g. whether there is any impact on patient safety, trial conduct or trial documentation).
Safety notifications and reporting pathways for non-therapeutic goods trials
Safety notifications for non-therapeutic goods trials
View safety notification summary table
- Type of event
- Who reports
- To whom
- When
- How
- Sponsor/Delegate
- The reviewing HREC (and all investigators participating in the study)
- As soon as possible and no later than 72 hours of the sponsor becoming aware of the USM
- SSI Notification Form; Sponsor’s template
- Sponsor/Delegate
- The reviewing HREC (and all investigators participating in the study)
- Within 15 days of the sponsor becoming aware of the SSI
- SSI Notification Form; Sponsor’s template
- Principal Investigator
- The RGO for the site where the event occurred
- As soon as possible and no later than 72 hours of the PI becoming aware of the SSI
- SSI Notification Form; Sponsor’s template
- Principal Investigator
- The RGO for the site where the event occurred
- Within 72 hours of the PI becoming aware of the event
- SUSAR/USADE/URSAE Notification Form
- Coordinating Principal Investigator or Sponsor/Delegate
- The reviewing HREC
- Annually (within the annual progress report)
- Annual Progress Report
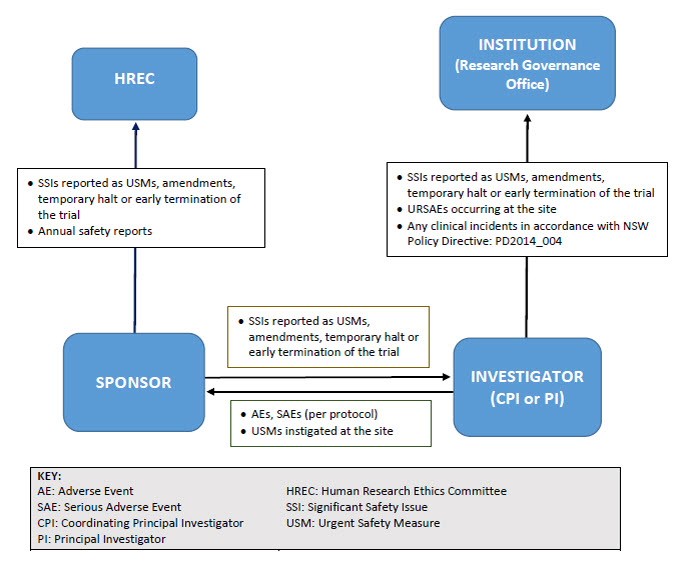
Reporting pathway for non-therapeutic goods trials
As illustrated below, sponsors may report directly to NSW Human Research Ethics Committees; however, they must ensure that all communications sent to the Committee adequately identify the trial and provide context in relation to the Committee’s role (e.g. whether there is any impact on patient safety, trial conduct or trial documentation).
Updated 8 months ago